
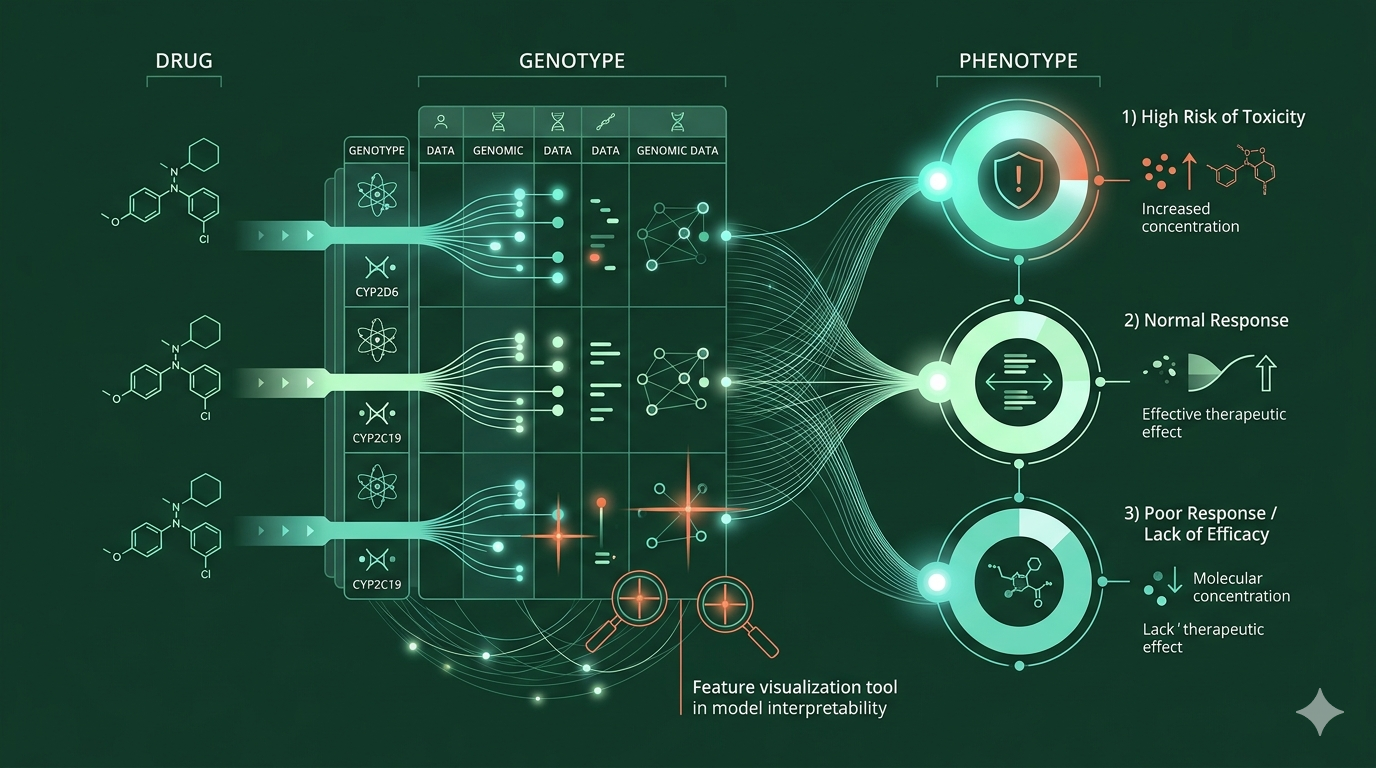
Představme si tři ženy, přibližně stejného věku, se stejnou diagnózou – mírnou depresivní a úzkostnou epizodou. Všechny tři dostaly od svého lékaře týž lék: paroxetin dvacet miligramů denně. Jedna z nich po dvou týdnech přestala spát. Měla nesnesitelnou nevolnost, pocit, že se jí zrychluje tep, přibírala, potila se, a nakonec přišla za lékařem s přesvědčením, že „na ni ty léky prostě nezabírají". Její sestra, která dostávala přesně totéž, po měsíci říkala opak: necítila žádné vedlejší účinky, ale ani žádné zlepšení. Nic. Třetí žena, z jiné rodiny, popisovala po šesti týdnech klidný, funkční život, vděčná za to, jak jednoduchá nakonec léčba byla.
Tři pacientky, tentýž lék, tatáž dávka, tři zcela odlišné příběhy. Jak je to možné?
Na část odpovědi stačí obecné znalosti klinické medicíny: záleží na věku, pohlaví, tělesné hmotnosti, jaterních a ledvinných funkcích, na tom, co pacient ještě současně užívá, a samozřejmě na tom, zda lék vůbec bere tak, jak se mu řekne. Další část vysvětluje samotná povaha deprese, která je heterogenní a nikdy se přesně neprojevuje u dvou lidí stejně. Ale významný, a pro moderní medicínu stále zásadnější, kus odpovědi je ukrytý v genetickém kódu každé z těch tří žen. Přesněji v tom, jak rychle jejich játra paroxetin odbourávají, jak jejich serotoninový systém reaguje na jeho přítomnost, a zda vůbec mají v dostatečně funkční podobě enzym, který má lék zpracovat.
Obor, který se touto variabilitou zabývá, se jmenuje farmakogenetika, a v modernější a širší podobě farmakogenomika. Existuje bezmála sedmdesát let. Většinu své historie byl okrajovým tématem klinické farmakologie, který zajímal hlavně akademiky. Teprve v posledních dvou dekádách se začíná prosazovat v běžné klinické praxi — pomalu, s mezerami, ale neodvratně.
Od favismu k prvním enzymům
Pozorování, že různí lidé reagují na stejnou látku odlišně, samo o sobě není nové. Pythagoras už v 6. století před naším letopočtem varoval své žáky, aby nejedli boby, protože pozorované případy hemolytické anemie u citlivých jedinců znal evidentně dost dobře na to, aby je byl schopen vypozorovat, že se nejedná o náhodný jev. Dnes víme, že šlo o favismus, tedy vrozený deficit enzymu glukóza-6-fosfát dehydrogenázy (G6PD). První systematický důkaz, že odpověď člověka na určitou chemickou látku se může mezi lidmi lišit a je dědičná, přišel ale až v roce 1932. Arthur Fox a Lawrence Snyder tehdy ukázali, že schopnost vnímat hořkou chuť látky jménem fenylthiokarbamid se mezi lidmi štěpí na „chutnače" a „nechutnače" podle klasických Mendelových zákonů.
Skutečné základy moderní farmakogenetiky se, nicméně, položily až ve čtyřicátých a padesátých letech 20. století, a to ve třech paralelních liniích výzkumu, které se zajímavým způsobem propojují s tématem jaterního metabolismu léků.
Za první mezník lze pokládat práci Alfa Alvinga z Chicaga, který v roce 1956 zkoumal, proč u některých amerických vojáků ve válce v Koreji vyvolává antimalarikum primaquin akutní hemolýzu, tj. — rozpad červených krvinek. Ukázalo se, že vojáci se zdravotními obtížemi měli deficit již zmíněné G6PD. Tato enzymopatie (onemocnění způsobená dědičnou nebo získanou poruchou funkce specifického enzymu, což vede k metabolickému bloku) je přitom ve světovém měřítku extrémně častá; v některých oblastech Afriky, Středomoří a jihovýchodní Asie ji má až třetina populace. Byl to první velký případ, kdy musela klinická praxe při běžném používání léku zohlednit lidskou genetickou variabilitu.
.
Druhým mezníkem byla práce Wernera Kalowa z Toronta. V roce 1957 popsal, že část pacientů po podání svalového relaxancia sukcinylcholinu během úvodní anestezie neprobouzela ze spánku hodiny místo obvyklých několika minut. Kalow ukázal, že za to může vrozená varianta enzymu butyrylcholinesterázy, který sukcinylcholin v krvi normálně rychle štěpí. Lidé se dvěma kopiemi atypické varianty (zhruba jeden člověk z 2 500 až 3 000) jej nestíhají zpracovat a po anestezii potřebují strávit hodiny na umělé plicní ventilace.
Třetí mezník přišel po tomv roce 1960. David Evans v roce 1960 popsal, že pacienti léčení pro tuberkulózu se při stejné dávce léku isoniazidu rozdělují na dvě skupiny: „rychlé acetylátory" a „pomalé acetylátory". Klíčovým rozdílem mezi pacienty bylo, jak rychlýe mají funkční enzym N‑acetyltransferázu 2. Pomalí měli v krvi vyšší koncentrace léku a častěji trpěli jeho nežádoucími účinky, zejména poškozením jater a periferních nervů. Rychlím naopak hrozilo, že jim lék „utekl" dřív, než stihl účinkovat, a tuberkulóza se u nich hůře léčila.
Obor v té chvíli dostal své jméno. Německý lékařský genetik Friedrich Vogel publikoval v roce 1959 v Heidelbergu článek, ve kterém poprvé použil termín „Pharmacogenetik". O dva roky dříve už koncepční rámec oboru načrtl v Americe Arno Motulsky, lékařský genetik německého původu působící na University of Washington v Seattlu, jehož odkaz dodnes ovlivňuje americké vzdělávání v klinické genetice. A v roce 1962 vydal Werner Kalow 231 stránkovou monografii Pharmacogenetics: Heredity and the Response to Drugs, první ucelenou knihu tohoto nového oboru.
Debrisochin, CYP2D6 a rozbité dogma
Skutečný zlom v ovlivnění širší medicíny přišel v roce 1975. Britský farmakolog Robert Smith ze St Mary's Hospital v Londýně testoval na sobě samém nové antihypertenzivum (léky snižující patologicky zvýšený krevní tlak) jménem debrisochin. Po běžné dávce, která všem ostatním dobrovolníkům v pilotní studii vyhovovala, sám upadl do takové hypotenze, že se musel položit a několik dní pracovat vleže. Spolu se svým doktorandem Alim Mahgoubem pak ukázal v přelomovém článku z roku 1977 v časopisu Lancet, že metabolismus debrisochinu v játrech se v populaci štěpí na dvě skupiny. Zhruba 5-10%10% Evropanů látku rozkládá dramaticky pomaleji než ostatní. Pro tyto jedince naevrhli oba vědci označení „poor metabolizers"(pomalí metabolizátoři)
Enzymem, který za tím stojí, se po dlouhém hledání ukázal být jeden z cytochromů P450 v játrech, který je kódován genem CYP2D6. A nejde o raritu, která by se dotýkala jen pacientů s debrisochinem. Tento jediný jaterní enzym se podílí na odbourávání přibližně čtvrtiny všech dnes předepisovaných léků. Pro naši diskusi o třech sestrách, které tak odlišně reagovaly na paroxetin, je to klíč.
Identifikace CYP2D6 změnila způsob, jakým lékaři začali uvažovat o dávkování. Dokud byla farmakogenetika raritní záležitostívzácnos, například u pacientů, kterým se po anestezii zastavil dech, nebo vojáků s hemolytickou anemií po antimalariku, mohl si celý obor dovolit působit jako zajímavost pro specialisty. S CYP2D6 to najednou přestalo platit. Variabilita se netýkala hrstky pacientů, nýbrž poměrně význačné části populace.
S rozvojem molekulární genetiky osmdesátých let se gen CYP2D6 sekvenoval, byly popsány desítky jeho funkčních variant a vznikla klasifikace, která se používá dodnes. Lidé, kteří mají dvě kopie nefunkčního genu, jsou pomalí metabolizátoři (= špatně odpovídají na léčbu). Ti s jednou funkční a jednou nefunkční alelou jsou intermediární. Většinu populace tvoří normální metabolizátoři. A konečně jsou lidé, kteří díky duplikacím funkčního genu mají aktivity enzymu nadbytek a kteří se označují jako ultrarychlí metabolizátoři (ultrarapid metabolizers). Každá z těchto kategorií může u téhož léku znamenat naprosto odlišný klinický obraz. Tam, kde je lék sám o sobě je účinnou látkou a játra jej jen pomalu odstraňují, způsobí u pomaléhoý metabolizátora vysoké koncentrace a toxicitu. Tam, kde je lék jen „proléčivem", které se teprve v játrech musí aktivovat na účinnou formu (a u antidepresiv a analgetik se to stává často) způsobí pomalý metabolizátor pravý opak, a sice nedostatek účinku.
Od farmakogenetiky k farmakogenomice
Projekt lidského genomu, dokončený v letech 2001 až 2003, posunul celý obor z úrovně „jeden gen, jeden lék" na úroveň celého genomu. Na konci prvního desetiletí 21. století začalo být prakticky proveditelné testovat desítky až stovky farmakogeneticky významných variant najednou, a to z jediného vzorku slin nebo krve. Změnilo se i jméno: místo o farmakogenetice se dnes stále častěji mluví o farmakogenomice, aby se zdůraznilo, že lék nepůsobí v kontextu jednoho genu, ale celých metabolických drah a jejich vzájemných interakcí.
S tímto posunem vznikla v klinické praxi i nová strategie: preemptivní testování. Myšlenka je jednoduchá. Pokud už jednou zjistíme, že pacient je například pomalý metabolizátor CYP2D6, tento údaj platí po celý jeho život. Nemá smysl čekat, až se dostane do nemocnice kvůli nečekané reakci na lék; vyplatí se výsledek uložit do zdravotnické dokumentace a použít ho pokaždé, když se v budoucnu bude rozhodovat o nové preskripci. Tento model se dnes v různé míře testuje v Nizozemí, Estonsku, Velké Británii a některých amerických zdravotnických systémech.
Ve stejné době se farmakogenomika poprvé dostala do oficiálních klinických doporučení. Dnes existují tři klíčové autority, na které se medicína v této oblasti odvolává. Americké (ale de facto mezinárodní) CPIC (Clinical Pharmacogenetics Implementation Consortiumv) ,), které vydává konkrétní návody, jak upravit dávku nebo zvolit jiný lék podle genotypu. Jeho evropskou obdobou je nizozemská skupina DPWG (Dutch Pharmacogenetics Working Group), jejíž doporučení se promítají do řady evropských guidelines. A konečně americký regulátor FDA vede veřejně dostupnou databázi farmakogenomické vyznaných genů, či konkrétních variant a jejich vztah k metabolizmu farmak. V současné době jde o stovky preparátů napříč obory.
Proč zrovna antidepresiva
Vrátíme se k třem sestrám z úvodu. Antidepresiva, a zvláště ze skupiny SSRI (selektivní inhibitory zpětného vychytávání serotoninu) a starší tricyklická antidepresiva (TCA) , jsou pro farmakogenomiku učebnicovým příkladem hned z několika důvodů.
Prvním důvodem je fakt, že se metabolizují právě přes enzymy CYP2D6 a CYP2C19, tedy dva nejlépe prozkoumané farmakogenetické cíle, jaké v klinické medicíně známe. Druhým je úzký terapeutický interval a obrovská individuální variabilita hladin v krvi při podání stejné dávky. U některých tricyklických antidepresiv se dokumentovaly dokonce třiceti‑ až padesátinásobné rozdíly mezi nejpomalejšími a nejrychlejšími metabolizátory. Rozdíl mezi „nic se neděje" a „pacient skončil na pohotovosti" tedy není rozdíl mezi různými léky, ale mezi různými pacienty na tomtéž léku.
Třetím důvodem je klinická realita. Odhaduje se, že asi třetina až dvě pětiny pacientů nezažije při prvním zvoleném antidepresivu dostatečný účinek. Další nezanedbatelné procento léčbu přeruší kvůli nežádoucím účinkům. U nějaké části těchto „neresponderů" a „intolerantů" přitom jde o lidi, u kterých by genetický test odhalil klinicky významnou variantuvariantu, a tudíž by pomohl lékaři se rozhodnout lépe rozhodnout hned napoprvé.
Čtvrtým důvodem je překvapivě vysoké zastoupení netypických fenotypů v populaci. V meta‑analýze z roku 2021, která zahrnula přes 336 000 lidí z celého světa, mělo nenormální fenotyp CYP2D6 přibližně 36 % populace a nenormální fenotyp CYP2C19 dokonce 62 %. Pokud tedy hodnotíme „průměrného pacienta" z hlediska těchto dvou enzymů, statisticky vzato je pravděpodobnější, že bude z farmakogenomického pohledu spíš atypický než typický. Jen si toho běžně nikdo nevšimne, protože se genotyp v ordinaci nezjišťuje.
Aktuální doporučení CPIC z roku 2023, která nahradila starší verzi z roku 2015, řeší jednotlivá antidepresiva docela konkrétně. Escitalopram a citalopram se odbourávají hlavně enzymem CYP2C19. U pomalých metabolizátorů tohoto enzymu CPIC doporučuje snížit dávku zhruba na polovinu nebo raději zvolit lék, který se přes CYP2C19 nemetabolizuje. U ultrarychlých metabolizátorů je zase na místě zvážit alternativu, protože hrozí, že lék jaterní enzym rozebere tak rychle, že se v mozku jeho terapeutické hladiny prostě nedosáhne. Podobně je to u sertralinu. Paroxetin a fluvoxamin jdou převážně metabolizovány pomocí CYP2D6 , pro který platí stejná logika: pomalý metabolizátor potřebuje nižší dávku, ultrarychlý jiný lék. U starších tricyklických antidepresiv, jako jsou amitriptylin nebo nortriptylin, se zohledňuje kombinovaný fenotyp obou enzymů a doporučení jsou podrobněji rozpracovaná už od roku 2013.
Co ukazují velké klinické studie
Mezi pacientskou intuicí „ten lék mi pomohl" a přísným vědeckým důkazem „tato intervence měřitelně zlepšuje klinické výsledky" bývá obykle v medicíně značný rozdíl. U farmakogenomiky antidepresiv tuto vzdálenost ilustrují tři důležité studie posledních let.
První z nich je GUIDED z roku 2019, randomizovaná kontrolovaná studie s tisícovkou sto šedesáti sedmi 1167 pacienty s rezistentní depresí. Polovina dostala léčbu vedenou komerčním panelovým testem GeneSight a druhá, polovina běžnou péči. Když se po osmi týdnech měřilo zlepšení depresivních příznaků pomocí standardní Hamiltonovy škály, rozdíl nebyl statisticky významný. Sekundární ukazatele (např. odpověď na léčbu a remise) vyšly numericky lépe ve skupině s genetickým testem, ale rozdíly byly nevelké a vyvolaly debatu o tom, zda jde o klinicky relevantní benefit, nebo spíše o statistický šum.
Druhá velká studie, PRIME Care z roku 2022, byla provedena v systému amerických vojenských nemocnic u veteránů US army. Zapojilo se téměř dva tisíce pacientů ve dvaadvaceti centrech. Ukázala dvě zajímavé věci. Za prvé: testování skutečně změnilo preskripční chování lékařů. Ve skupině s dostupným genetickým výsledkem bylo podstatně méně situací, kdy pacient dostal lék s „podstatnou" genově ‑ lékovou interakcí (asi 11 % proti 20 % v kontrole). Za druhé, tato změna se jen mírně promítla do remise depresivních příznaků (maximálně o pět procentních bodů ve dvanáctém týdnu, a ve 24. týdnu už rozdíl nebyl statisticky významný). Test tedy ovlivňuje to, co lékaři dělají; otázkou zůstává, jak moc to pacientům reálně pomáhá.
Třetí a pro Evropu nejdůležitější studií je PREPARE, publikovaná v Lancetu v roce 2023. Šlo o velkou implementační studii s dvanáctigenovým panelem u téměř sedmi tisíc pacientů v sedmi evropských zemích (Nizozemí, Španělsku, Itálii, Rakousku, Řecku, Slovinsku a Velké Británii). Studie měla podobu takzvané crossover cluster‑randomizovaná studie, která se snažila co nejvíce přiblížit reálné klinické praxi. U pacientů, jejichž preskripce byla vedena genotypem, byl výskyt klinicky významných nežádoucích lékových reakcí o 30 % nižší než u pacientů v běžné péči. Na rozdíl od GUIDED a PRIME Care zde tedy byla zjištěna robustní, statisticky velmi přesvědčivá redukce nežádoucích účinků. Později se sice ukázalo, že studie měla řadu nedostatků. Nebyla například zaslepená, přibližně desetina pacientů vypadla ze sledování a kauzalitu nežádoucích účinků hodnotil nezaslepený posuzovatel. Nezávislá kontrola, kterou provedli editoři Lancetu v následném komentáři, ukázala jen mírnou shodu mezi hodnotiteli. Nejde tedy o důkaz tak tvrdý, jaký by si čtenář mohl přát, ale zároveň jde o dosud nejpřesvědčivější evropská data ve prospěch plošného genotypování.
Když tyto tři studie shrneme, vyjde z toho celkem zřetelný obraz: farmakogenomické testování zásadně mění preskripci a pravděpodobně snižuje nežádoucí účinky; jeho dopad na samotnou účinnost léčby (například na dosažení remise deprese) je měřitelný jen omezeně a liší se případ od případu. Farmakogenomika nemá smysl jako plošné opatření pro celou populaci, ale má jasný smysl tam, kde už pacient s léčbou neúspěšně bojuje, má netoleranci, plánuje se dlouhodobá polymedikace, nebo je přítomen lék s úzkým terapeutickým rozpětím.
Když genetika zachraňuje životy: příklady z jiných oborů
Antidepresiva jsou vděčné učebnicové téma, ale skutečně dramatické případy farmakogenomiky leží mimo psychiatrii. Tam, kde neodhalená varianta v genomu znamená rozdíl mezi životem a smrtí pacienta.
Kodein je přírodní opiát (alkaloid opia) používaný k léčbě suchého, dráždivého kašle a středně silné bolesti. Sám o sobě bolest neutišuje, jelikož se játrech musí přeměnit enzymem CYP2D6 na morfin, a právě ten působí analgeticky. U pomalého metabolizátora je tedy analgezie minimální. U ultrarychlého metabolizátora se ovšem kodein proměňuje na morfin tak intenzivně, že koncentrace morfinu v krvi dosáhne toxických hodnot. V letech 2009–2013 bylo popsáno několik tragických případů u dětí, kterým byl kodein podán rutinně po operaci krčních mandlí, přičemž některé z nich následně zemřely na dechovou depresi. FDA následně zavedla tzv. black box warning (nejvyšší úroveň výstrahy pro léčivo v registru FDA - u léčiva existuje důkaz o závažném, potenciálně život ohrožujícím nežádoucím účinku daného léku) a posléze kontraindikaci pro pediatrii po tonzilektomii a adenoidektomii. Zmínku zaslouží i případ kojeného novorozence, který zemřel, protože jeho matka byla ultrarychlý metabolizátor a morfin procházel do mateřského mléka v nebezpečných koncentracích.
Dalším případem se závažným klinickým dopadem je antiretrovirotikum abakavir, používané v léčbě HIV, které vyvolává u 5-10% pacientů těžkou hypersenzitivní reakci, která při opakovaném podání může být smrtelná. Už v roce 2002 bylo popsáno, že tato reakce je téměř výhradně vázaná na přítomnost alely HLA‑B*57:01. Prospektivní studie PREDICT‑1 publikovaná v New England Journal of Medicine v roce 2008 ukázala, že pokud se před zahájením léčby provede genetický test, výskyt imunologicky potvrzených hypersenzitivních reakcí klesne z 2,7 % na nulu. Screening na HLA‑B*57:01 je dnes mezinárodně přijatým standardem a bez něj se abakavir fakticky nepředepisuje.
Z onkologie jsou popsány případy negativních účinků tiopurinů TPMT nebo NUDT15. Azathioprin, 6‑merkaptopurin a thioguanin jsou léky používané u dětské akutní lymfoblastické leukemie, autoimunitních onemocnění nebo po transplantaci orgánů. U lidí s vrozeně nízkou aktivitou enzymu TPMT nebo příbuzného NUDT15 hrozí po běžné dávce fatální útlum funkcí kostní dřeně, kdy pacient může během několika týdnů zemřít na infekci, protože nemá bílé krvinky. CPIC doporučuje u homozygotních pomalých metabolizátorů (vrozeně zcela nefunkční enzym) snížit dávku o 80-90% nebo zvolit jiný lék; u mnohem častějších intermediárních metabolizátorů se doporučuje dávku redukovat přibližně o 20-70% podle konkrétního léku a indikace. V dětské onkologii je testování TPMT už několik let zcela rutinní.
Fluoropyrimidiny (5‑fluorouracil, capecitabin) jsou pilíř chemoterapie gastrointestinálních a dalších solidních nádorů. Přibližně 3–8% evropské populace nese patogenní variantu v některém z klinicky relevantních míst genu DPYD, který kóduje lék degradující enzym DPD. Pacienti s deficitem DPD mohou po běžné dávce zemřít na těžkou toxicitu v důsledku: zvracení, průjemu, rozpadu sliznic a , útlumu funkcí kostní dřeně. Homozygoti s kompletním deficitem mají riziko úmrtí několik procent; heterozygoti riziko těžké toxicity nad padesát procent. Evropská léková agentura doporučuje od roku 2020 prospektivní testování buď genotypu DPYD, nebo fenotypu (měřením hladiny uracilu v plazmě) před zahájením léčby. Ve Francii a Belgii je to závazné; v České republice se používá zatím jen ve vybraných onkologických centrech.
Warfarin je známý antikoagulant, u kterého rozdíl mezi účinnou a nežádoucí dávkou způsobujíci krvácivost, který může být mezi dvěma pacienty desetinásobný. Kombinace klinických údajů s genotypem dvou genů (CYP2C9 a VKORC1) vysvětlí zhruba polovinu této variability. V éře přímých perorálních antikoagulancií (DOAC) se sice význam warfarinu postupně zmenšuje, ale tam, kde se stále používá, je genetická informace při hledání stabilní dávky užitečná.
Velikost problému: proč to vůbec řešíme
Nežádoucí lékové reakce (ADR, adverse drug reactions) nejsou okrajový zdravotnický problém. Systematický přehled publikovaný v časopise Drug Safety v roce 2015 uvádí, že přibližně 3,6 % všech hospitalizací v Evropě je přímo způsobeno nežádoucí lékovou reakcí, a dalších zhruba deset procent hospitalizovaných pacientů takovou reakci zažije během pobytu v nemocnici. Odhady mortality v Evropské unii se pohybují mezi čtyřiceti tisíci a téměř dvěma sty tisíci úmrtí ročně. Šířka toho intervalu sama o sobě ukazuje, jak nespolehlivě se tyto věci odhadují. V americkém kontextu klasifikovala American Society of Pharmacovigilance v roce 2025 nežádoucí lékové události jako třetí nejčastější příčinu úmrtí, s odhadem čtvrt milionu úmrtí ročně. Metodicky je to číslo sporné, ale vyjadřuje to, o čem lékaři v nemocnicích vědí ze své klinické praxe – že nežádoucí účinky léků jsou velmi reálný a velmi rozšířený problém.
Jak jsme na tom v Česku
České zdravotnictví farmakogenomiku formálně uznává, ale v klinické praxi ji využívá spíše okrajově. Testování genů CYP2D6 a CYP2C19 je v České republice hrazené z veřejného zdravotního pojištění, ale pouze tehdy, když vyšetření indikuje klinický genetik. To znamená, že většina běžných lékařů k němu reálně nemá přímý přístup, protože musí pacienta nejprve odeslat ke genetikovi, který vyšetření indikuje a hradí. Není to neschůdná cesta, ale v praxi vytváří bariéru, která vede k tomu, že testování se téměř nepoužívá tam, kde by mohlo pacientům pomoci nejvíc, a sice v ordinacích primární péče. Retrospektivní analýza ve Fakultní nemocnici Brno‑Bohunice za období 1997–2019 ukázala, že v hodnoceném souboru testováním prošlo méně než procento pacientů s depresivní epizodou během prvního roku po stanovení diagnózy. Mezi pacienty, kteří testem prošli, přitom asi šedesát procent dostalo do roka jiné antidepresivum než to, kterým začalo, což naznačuje, že test skutečně ovlivňuje klinické rozhodování.
Paralelně existuje trh komerčně nabízených panelů. Tyto testy si obvykle pacienti hradí sami, v cenách řádově tisíců až desítek tisíc korun. VZP ve svém oficiálním metodickém stanovisku k laboratorním genetickým vyšetřením upozorňuje, že většina těchto rozšířených panelů není hrazena plošně, a indikaci musí provést klinický genetik na základě klinického vyšetření, tedy ne automaticky na přání pacienta.
Paradoxní situace českého prostředí spočívá v tom, že dostupnost špičkových sekvenačních technologií celogenomového nebo celoexomového sekvenování je v tuzemsku relativně vysoká (včetně komerčních laboratoří jako Sophgena), zatímco systematické využívání farmakogenomických dat v běžné preskripční praxi zaostává. Technologicky máme na co navázat, chybí zde spíš infrastruktura pro propojení výsledků se zdravotnickými informačními systémy a vzdělávání lékařů primární péče.
Co farmakogenomika (zatím) nedovede
Bylo by nezodpovědné skončit článek tím, že bych čtenáři nalhával představu, že genetický test vyřeší všechno. Farmakogenomika má jasné a reálné limity a je poctivější je pojmenovat.
I u nejlépe prostudovaných léků, jakým je warfarin, vysvětluje genotyp jen část variability účinku. Zbytek jde na vrub věku, funkce jater a ledvin, současně užívané medikace, diety, kouření, tělesné hmotnosti, epigenetických faktorů a samozřejmě adherence. Genetika je jedna, silná, ale nikoli jediná proměnná.
Další limit se jmenuje fenokonverze. Geneticky normální metabolizátor, který začne užívat silný inhibitor svého vlastního enzymu (a některá antidepresiva takovými inhibitory jsou — paroxetin například silně inhibuje CYP2D6), se najednou chová, jako by byl pomalým metabolizátorem. Jeho genotyp se samozřejmě nezměnil; změnilo se jeho chemické prostředí. Genetický test tuto situaci nedokáže předpovědět; je to práce klinického farmakologa.
Třetí limit je etnický. Převážná většina velkých farmakogenomických studií byla provedena na populacích evropského původu. Spektrum farmakogenomicky významných variant může být ale u afrických, asijských a indigenních populací výrazně odlišné, a proto extrapolace evropských doporučení na jiné etnické kontexty není vždy validní.
Čtvrtý limit technický. Farmakogenomické testy mají svou pozitivní i negativní prediktivní hodnotu, které nejsou stoprocentní. Například genetický test pro gen DPYD zachytí zhruba dvacet až třicet procent časných těžkých toxicit 5‑fluorouracilu, zatímco zbylé mají jiné příčiny, které nevidíme. Pozitivní test neznamená jistou toxicitu, negativní neznamená její vyloučení.
Nelze také pominout finanční rámec. Nákladová efektivita záleží na tom, kdo je testován a na co. Plošné testování celé populace má smysl jen tehdy, pokud testovaná varianta má vysokou prevalenci, významné klinické dopady a existuje alternativní léčebná strategie. U cíleného testování (u pacientů zahajujících chemoterapii, u rezistentní deprese, u vybraných kardiologických indikací ) jsou data o nákladové efektivitě mnohem přesvědčivější.
Výhled do budoucna
Pokud se na vývoj v posledních dekádách podíváme s určitým odstupem, vynořují se tři trendy, které budou pravděpodobně definovat další desetiletí.
Prvním je preemptivní panelové testování integrované do elektronické dokumentace pacienta. Jakmile člověk jednou má farmakogenomický profil, tento profil platí doživotně ( pomíjíme-li vliv epigenetiky). Vkládání tohoto výsledku do zdravotnického informačního systému a napojení na klinické rozhodovací systémy tak, aby se lékaři při každé nové preskripci automaticky zobrazila varovná nebo doporučující zpráva, což je model, který se dnes v různé míře testuje v nizozemském, estonském i americkém zdravotnictví.
Druhým trendem je integrace s polygenními skóre. Klasická farmakogenomika pracuje s relativně mendelovskými variantami velkého efektu (např. CYP2D6, HLA‑B*57:01). Polygenní skóre naproti tomu kombinují desítky až tisíce variant s malým individuálním efektem. Dnes už se testují polygenní prediktory odpovědi na antidepresiva, statiny nebo metformin. Jejich klinická validita je ale zatím předběžná; je to oblast, kde se očekává hodně, ale zatím není nic, o co by se dalo v ordinaci bez výhrad opřít.
Třetím trendem je přesun od cílených panelů k celogenomovému sekvenování. V okamžiku, kdy pacient podstoupí celogenomové sekvenování z jiného diagnostického důvodu, je farmakogenomická informace v podstatě vedlejším produktem bez dodatečných nákladů. Dosud jsme si kladli otázku, zda má smysl pacientovi farmakogenomický test indikovat. Budoucnost bude čím dál častěji klást otázku obrácenou: jak nejlépe interpretovat farmakogenomická data, která už stejně máme. V českém kontextu, kde se celogenomové sekvenování v klinice postupně prosazuje, je to otázka, kterou bude nutné řešit systémově od laboratorních standardů interpretace přes strukturu zpráv pro lékaře až po vzdělávání praktiků.
Reálný krok, který by v české praxi mohl přinést největší okamžitý užitek, přitom není žádný technologický. Je jím integrace stávajících, již hrazených testů do běžných klinických algoritmů, zejména v psychiatrii, kardiologii a onkologii, a systematické vzdělávání lékařů prvního kontaktu. Jinak bude dál platit zkušenost, se kterou tento článek začíná: „Paroxetin mi prostě nesedl." Možná ne proto, že by byl špatným lékem. Možná proto, že byl dobrým lékem pro někoho jiného.
Zdroje
American Society of Pharmacovigilance. Adverse drug events now the third leading cause of death (press release, 2025).
https://www.prnewswire.com/news-releases/americas-silent-epidemic-worsens-adverse-drug-events-now-the-third-leading-cause-of-death-302408765.html
Amstutz U, Henricks LM, Offer SM, et al. CPIC Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing.
https://pmc.ncbi.nlm.nih.gov/articles/PMC9610761/
Bousman CA, Stevenson JM, Ramsey LB, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6, CYP2C19, CYP2B6, SLC6A4, and HTR2A Genotypes and Serotonin Reuptake Inhibitor Antidepressants. Clinical Pharmacology & Therapeutics (2023).
https://pubmed.ncbi.nlm.nih.gov/37032427/
Bouvy JC, De Bruin ML, Koopmanschap MA. Epidemiology of Adverse Drug Reactions in Europe: a Review of Recent Observational Studies. Drug Safety 38, 437–453 (2015).
https://pmc.ncbi.nlm.nih.gov/articles/PMC4412588/
Češková E, Šilhán P. Současnost a budoucnost farmakogenetického testování se zaměřením na farmakoterapii deprese. Psychiatrie pro praxi 25(2), 2024.
https://www.psychiatriepropraxi.cz/pdfs/psy/2024/02/02.pdf
Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens–Johnson syndrome. Nature 428, 486 (2004).
https://www.nature.com/articles/428486a
Clinical Pharmacogenetics Implementation Consortium (CPIC) — Guidelines.
https://cpicpgx.org/
Comment / correspondence on PREPARE trial methodology.
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(23)00855-3/fulltext
CPIC. Guideline for SSRI antidepressants — full PDF (2023).
https://files.cpicpgx.org/data/guideline/publication/serotonin_reuptake_inhibitor_antidepressants/2023/37032427.pdf
Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for codeine therapy in the context of CYP2D6 genotype (NCBI Bookshelf).
https://www.ncbi.nlm.nih.gov/books/NBK100662/
GHC Genetics. Jak může genetika pomoci pacientům s duševními chorobami — komentář k panelům typu Pharmagen.
https://www.ghcgenetics.cz/genopedie/jak-muze-genetika-pomoci-pacientum-s-dusevnimi-chorobami
Greden JF, Parikh SV, Rothschild AJ, et al. Impact of pharmacogenomics on clinical outcomes in major depressive disorder in the trial. Journal of Psychiatric Research 111, 59–67 (2019).
https://pubmed.ncbi.nlm.nih.gov/30677646
Kalow W. Pharmacogenetics and pharmacogenomics: origin, status, and the hope for personalized medicine. The Pharmacogenomics Journal 6, 162–165 (2006).
https://pmc.ncbi.nlm.nih.gov/articles/PMC2014592/
Mahgoub A, Idle JR, Dring LG, Lancaster R, Smith RL. Polymorphic hydroxylation of debrisoquine in man. The Lancet 310 (8038), 584–586 (1977).
https://pubmed.ncbi.nlm.nih.gov/71400/
Mallal S, Phillips E, Carosi G, et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. New England Journal of Medicine 358, 568–579 (2008).
https://pubmed.ncbi.nlm.nih.gov/18256392/
McInnes G, Lavertu A, Sangkuhl K, et al. Pharmacogenetics at scale: an analysis of the UK Biobank.
https://pmc.ncbi.nlm.nih.gov/articles/PMC7904867/
Meyer UA. Pharmacogenetics — five decades of therapeutic lessons from genetic diversity. Nature Reviews Genetics 5, 669–676 (2004).
https://pubmed.ncbi.nlm.nih.gov/15372089/
Motulsky AG, Qi M. Pharmacogenetics, pharmacogenomics and ecogenetics.
https://www.thieme-connect.com/products/ejournals/pdf/10.1055/a-0979-2322.pdf
Oslin DW, Lynch KG, Shih MC, et al. Effect of Pharmacogenomic Testing for Drug-Gene Interactions on Medication Selection and Remission of Symptoms in Major Depressive Disorder: The PRIME Care Randomized Clinical Trial. JAMA (2022).
https://jamanetwork.com/journals/jama/fullarticle/2794053
Pirmohamed M. Pharmacogenomics: current status and future perspectives.
https://www.frontiersin.org/journals/pharmacology/articles/10.3389/fphar.2021.638885/full
Relling MV, Schwab M, Whirl-Carrillo M, et al. CPIC Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018/2026 update.
https://files.cpicpgx.org/data/guideline/publication/thiopurines/2026/41618934.pdf
Swen JJ, Nijenhuis M, de Boer A, et al. Pharmacogenetics: from bench to byte — an update of guidelines. DPWG.
https://pmc.ncbi.nlm.nih.gov/articles/PMC8971318/
Swen JJ, van der Wouden CH, Manson LE, et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: an open-label, multicentre, controlled, cluster-randomised crossover implementation study (PREPARE). The Lancet 401, 347–356 (2023).
https://www.thelancet.com/journals/lancet/article/PIIS0140-6736(22)01841-4/abstract
U.S. Food and Drug Administration. Table of Pharmacogenomic Biomarkers in Drug Labeling.
https://www.fda.gov/drugs/science-and-research-drugs/table-pharmacogenomic-biomarkers-drug-labeling
Všeobecná zdravotní pojišťovna ČR. Informace VZP ČR k indikaci a vykazování laboratorních genetických vyšetření (odbornost 816).
https://www.vzp.cz/poskytovatele/informace-pro-praxi/vykazovani-a-uhrady/informace-vzp-cr-k-indikaci-a-vykazovani-laboratornich-genetickych-vysetreni-odbornost-816





