
Váš genetický test vám řekl, že je vše v pořádku. Ale analyzoval jen 1–2 % vašeho genomu. Zbývajících 98–99 % zůstalo nepřečtených."
Tato věta může znít provokativně. Přesto je věcně přesná — a je to jeden z nejdůležitějších faktů, které byste měli znát dříve, než si necháte udělat jakoukoli genetickou analýzu.
Genom není jedno území. Je to kontinent.
Lidský genom obsahuje přibližně 3,2 miliardy párů nukleotidů, základních stavebních kamenů DNA. Genetické varianty, tedy změny v sekvenci těchto nukleotidů, mohou způsobit onemocnění a navzájem se liší nejen polohou, ale i povahou. Nestačí tedy jen „přečíst nějakou část genomu" klíčové je, co přesně analýza zachytí a co přehlédne.
Z klinického hlediska rozeznáváme sedm hlavních kategorií variant:
- SNV (jednobodové záměny) záměna jednoho nukleotidu za jiný
- Malé inzerce a delece (indely) vložení nebo ztráta jednotek až desítek nukleotidů
- Změny počtu kopií (CNV) úseky DNA přítomné ve více nebo méně kopiích, než je obvyklé
- Strukturní varianty (SV) inverze, translokace a delece větších chromozomálních úseků
- Expanze krátkých tandemových repetic (STR) rozmnožení krátkých DNA motivů opakujících se za sebou
- Mitochondriální varianty varianty v mitochondriálním genomu, mimo jaderný genom
- Mozaikové varianty varianty přítomné jen v části buněk organismu
Každý typ má jiné klinické dopady a každá metoda ho zachycuje jinak. Tady začíná příběh o tom, proč na výběru testu skutečně záleží.
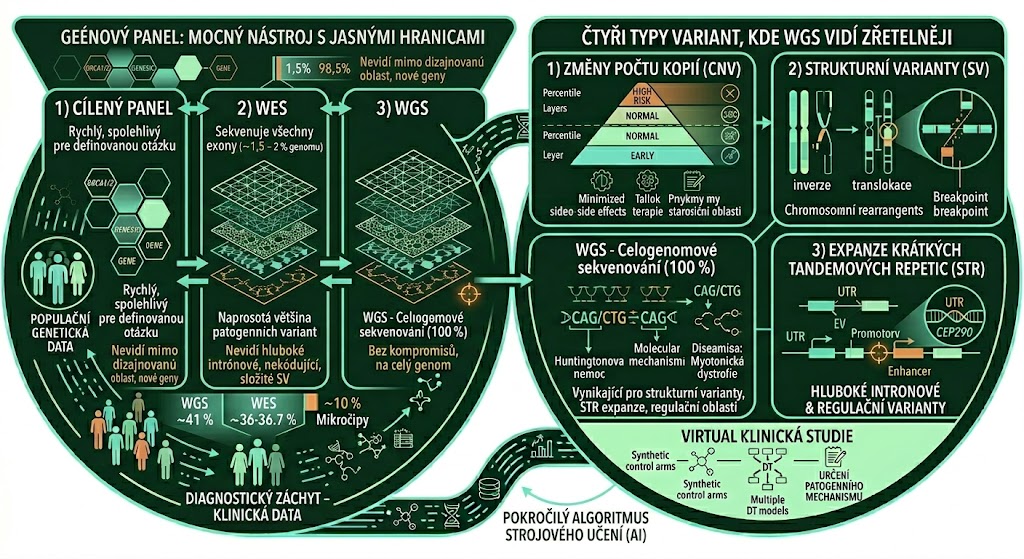
Panel: mocný nástroj s jasnými hranicemi
Genový panel je soubor předem definovaných genů analyzovaných cílenou metodou. Pokud například testujete riziko dědičného karcinomu prsu, panel pro geny BRCA1 a BRCA2 je rychlý, cenově dostupný a pro danou otázku spolehlivý.
Jenže panel má základní omezení: zachytí jen to, na co byl navržen.
- Patogenní varianta v genu, který do panelu nebyl zařazen? Neviditelná.
- Předem nepopsaná varianta v regulační oblasti genu nebo v jeho nekódující části? Neviditelná.
- Nový gen, jehož asociace s onemocněním byla popsána teprve loni? Neviditelný dokud panel neaktualizujete.
Panely jsou správnou volbou tam, kde je klinická otázka jasně definovaná a cílové geny jsou dobře charakterizovány. Jakmile se otázka rozšíří, nebo se hledá příčina dosud nediagnostikovaného onemocnění, panel naráží na své limity.
WES: velký skok, ale stále jen 1–2 % genomu
Celoexomové sekvenování (whole-exome sequencing, WES) představuje logický posun: místo omezeného panelu genů analyzuje všechny kódující oblasti genomu, tedy všechny exony všech genů. Exony jsou části genu nesoucí informaci pro stavbu proteinu; oddělují je introny, které představují nekódující mezisekvence v genu. Při přepisu genetické informace z DNA (transkripce) vzniká nejprve nezralá RNA obsahující oba typy sekvencí. Teprve po sestřihu (splicing) jsou introny odstraněny a zbylé exony tvoří zralou messengerovou RNA (mRNA). Ta se skládá ze tří částí: nepřekládaných oblastí na obou koncích (untranslated regions, UTR) a kódující oblasti uprostřed (coding sequence, CDS), která jako jediná nese instrukce pro syntézu proteinu.
Proč je to pokrok? Protože WES sekvenuje přibližně 20 000 genů najednou a naprostá většina dosud popsaných patogenních variant leží právě v kódujících oblastech genomu. WES proto dokáže identifikovat příčinu vzácného onemocnění i tehdy, kdy cílený panel selhal.
Jenže exom představuje jen přibližně 1–2 % celého genomu. Moderní klinické platformy na bázi WES, tzv. klinický exom (clinical WES, cWES), toto částečně kompenzují rozšířením záběru do nepřekládaných oblastí (untranslated regions, UTR) a do sestřihových míst v blízkosti exon-intronových hranic. Toto rozšíření se však zpravidla vztahuje jen na vybrané geny, u nichž jsou patogenní varianty v těchto oblastech klinicky dobře popsány, nikoli plošně na celý exom. Hluboké intronové varianty vzdálené stovky až tisíce bází od nejbližšího exonu cWES též nepokryje.
A co se skrývá ve zbývajících 98–99 % genomu? Původně se tyto rozsáhlé oblasti označovaly jako „junk DNA" předpokládalo se, že nemají žádnou funkci. Dnes víme, že to tak není. Nachází se zde:
- Regulační sekvence (promotory, enhancery) řídící, kdy a kde se exprese genů zapíná a vypíná
- Centromery a telomery zajišťující správné dělení a ochranu chromozomů
- Nekódující RNA (lncRNA, miRNA a další) hrající klíčové role v regulaci genové exprese
- Strukturní a scaffold oblasti udržující prostorovou organizaci chromatinu
Díky sekvenování celého genomu do tohoto prostoru konečně vidíme a katalog variant v těchto oblastech genomu s prokázaným klinickým dopadem se rozrůstá. Výsledek? Napříč různými skupinami onemocnění se diagnostický záchyt WES pohybuje v rozsahu 5–50 %. Tento extrémně široký interval odráží problém pozorovaný v klinické praxi: pro některé diagnózy je WES vynikající, pro jiné je vysoce neefektivní.
Čtyři typy variant, kde WGS vidí zřetelněji
Celogenomové sekvenování (whole-genome sequencing, WGS) analýzu rozšiřuje na celý genom, bez cílení na jednu konkrétní oblasti. To není jen kvantitativní rozdíl. Jde o kvalitativně odlišnou schopnost zachytit třídy variant, které může WES přehlédnout.
- Změny počtu kopií (CNV)
Jsou úseky DNA přítomné v jiném počtu kopií, než je u zdravých jedinců obvyklé. Buď je kopií navíc, nebo naopak chybí. Onemocnění mohou způsobit dvěma hlavními způsoby: buď narušením struktury samotného genu, tedy přerušením jeho kódující sekvence nebo sestřihových signálů, nebo změnou genové dávky, kdy se v důsledku odlišného počtu kopií genu či jeho regulačních oblastí mění množství produkovaného proteinu, ačkoli jeho sekvence může být zcela v pořádku
WES dokáže detekovat řadu CNV díky optimalizaci platformy a pomocí pokročilých bioinformatických nástrojů. V praxi to má ale výrazná omezení. Protože WES cíleně obohacuje jen vybrané oblasti genomu, je výsledné pokrytí cílených oblastí nerovnoměrné, a právě tato nerovnoměrnost komplikuje spolehlivou detekci malých CNV, zpravidla těch zahrnujících méně než přibližně 10 exonů. Velké chromozomální přestavby, jako je delece 22q11.2 u DiGeorge syndromu nebo delece 7q11.23 u Williamsova syndromu, WES obvykle zachytí. Menší, klinicky závažné CNV však spolehlivě identifikovat nedokáže a přesné určení míst zlomů (breakpoints), důležité pro klinickou interpretaci, WES v zásadě neumožňuje. WGS naproti tomu poskytuje rovnoměrné pokrytí celého genomu, které detekci CNV zásadně zlepšuje, jak v citlivosti, tak v rozlišení.
- Strukturní varianty (SV)
Vedle změn počtu kopií existují strukturní varianty v užším smyslu inverze, translokace a komplexní chromozomové přestavby, při nichž se genomové úseky otočí, přemístí nebo přeskupí, aniž by nutně docházelo ke ztrátě či zisku genetického materiálu. WES zachytí ty z nich, jejichž místa zlomů leží přímo v kódujících oblastech. Systematická detekce tohoto typu variant ale není součástí standardního WES protokolu ve většině laboratoří a závisí na dostupnosti pokročilé bioinformatické infrastruktury.
Ilustrativním příkladem jsou kryptické translokace, tedy chromozomové přestavby, které jsou příliš malé na to, aby je zachytil standardní karyotyp, a jejichž místa zlomů leží mimo kódující oblasti, takže WES je minul. U pacientů s nevysvětlitelnou vývojovou poruchou, opakovanými potraty nebo poruchou plodnosti s negativním karyotypem a negativním WES právě WGS tyto přestavby dokáže odhalit, bez nutnosti předem vědět, kde hledat.
- Expanze krátkých tandemových repetic (STR)
Některé části genomu, včetně oblastí uvnitř genů i mimo ně, obsahují krátké opakující se sekvenční motivy DNA, tzv. tandemové repetice. V případě STR se jedna o opakující se motivy dlouhé 1-6 nukleotidů. Existují i dlouhé tandemové repetice (n= 7-100) a minisality (n>100), ale klinicky nejrelevantnější expanze jsou způsobeny STR. Pokud se tento motiv zopakuje příliš mnohokrát, v řádech stovek až tisíců, výsledkem může být závažné onemocnění. Mechanismy jsou různé: expanze může umlčet gen (transkripční silencování), vytvářet toxické RNA molekuly sekvestrující důležité proteiny, nebo narušovat sestřih výsledné RNA.
Detekce STR je nejčistějším příkladem technologické nadřazenosti WGS nad WES. Není to otázka úpravy dizajnu platformy nebo bioinformatického postupu, ale o technologickou limitaci sekvenování krátkými čteními (short-read sequencing, technologický přístup sekvenačních platforem Illumina, Element Bioscience, MGI a dalších). Krátká čtení nedokážou spolehlivě mapovat dlouhé oblasti opakujících se sekvencí a PCR amplifikace při přípravě WES knihovny situaci ještě zhoršuje. Opakující se motivy jsou amplifikovány s nižší efektivitou a ve výsledných datech jsou podhodnoceny. WES zde tedy neselhává graduálně, ale principiálně.
STR jsou přitom příčinou více než 40 neurologických a neuromuskulárních onemocnění, jakými jsou například:
- Huntingtonova nemoc (expanze CAG v genu HTT)
- Myotonická dystrofie typ 1 (expanze CTG v genu DMPK)
- Friedreichova ataxie (expanze GAA v intronu genu FXN)
- Syndrom fragilního X (expanze CGG v genu FMR1)
WGS tuto bariéru posouvá. Bez cílení na konkrétní oblasti dokáže identifikovat vybrané STR jako screeningový nástroj napříč celým genomem najednou, bez nutnosti testovat každou diagnózu zvlášť. Pozitivní nález je pak standardně potvrzován ortogonální metodou, například specifickým typem PCR testu, který expanzi přesně kvantifikuje. Pro nejkomplexnější případy, kde je délka expanze kritická pro prognózu nebo kde WGS přesto selhává, představuje sekvenování dlouhými čteními (long-read sequencing, např. Oxford Nanopore nebo PacBio) zatím nejsilnější dostupný nástroj, protože dokáže sekvenovat i velmi rozsáhlé repetitivní úseky v jediném čtení.
- Hluboké intronové a regulační varianty
Geny nepracují izolovaně. Jejich aktivitu řídí regulační oblasti, jakými jsou promotory, enhancery a sestřihové sekvence, které leží mimo exony a jsou pro WES systematicky nedostupné.
Klinický dopad ilustruje dobře zdokumentovaný příklad: varianta c.2991+1655A>G v genu CEP290 leží přibližně 1,7 kb od nejbližšího exonu a způsobuje těžkou formu Leberovy vrozené amaurózy. WES tuto oblast nepokryl. WGS ji zachytí a ve studii na 33 pacientech, u nichž standardní NGS selhalo, vyřešil diagnózu u třetiny z nich.
Právě tyto varianty tvoří nezanedbatelnou část nevyřešených diagnóz. Ve studii na 122 pacientech s negativním výsledkem předchozího testování WGS vyřešilo diagnózu u 35 % z nich. Čím to je? Strukturní, sestřihové a hluboké intronové varianty, tedy přesně ty kategorie popsané v této kapitole, se podílely na téměř polovině všech nově nalezených diagnóz.
Proč na tom v praxi záleží
Mohlo by se zdát, že výše popsané kategorie variant jsou specializovanou záležitostí vzácných onemocnění a jsou relevantní jen pro úzké skupiny pacientů. Ale opak je pravdou. Všechny čtyři typy variant popsaných v předchozí kapitole mají jedno společné: standardní NGS metodiky je můžou přehlédnout nebo je nedokážou identifikovat vůbec. A právě to se odráží v číslech. Ve studii na 122 pacientech s negativním výsledkem předchozího panelového nebo exomového testování WGS vyřešilo diagnózu u 35 % z nich — přičemž strukturní, sestřihové a hluboké intronové varianty se podílely na téměř polovině všech nově nalezených diagnóz.
Nejde o ojedinělý výsledek. Metaanalýza zahrnující 20 068 dětí s podezřením na genetické onemocnění zjistila diagnostický záchyt 41 % pro WGS, oproti 36 % pro WES a pouhých 10 % pro mirkočipové platformy. Novější prospektivní srovnávací studie z roku 2025 potvrdila podobný trend: trio-WGS dosáhlo záchytu 40 % oproti 36,7 % pro standardní exomový přístup, přičemž klíčový přínos WGS spočíval právě v zachycení hlubokých intronových, nekódujících a malých CNV variant.
Procentuální rozdíly se mohou zdát malé ale pokud se na ně podíváme z perspektivy skutečných lidí ve zdravotnickém systému, jde o stovky až tisíce pacientů, kteří díky WGS konečně po zdlouhavém diagnostickém procesu dostávají odpověď.
A co preventivní genetika? Zde je situace méně jednoznačná. Systematický přehled 130 studií potvrzuje nákladovou efektivitu WGS v diagnostickém kontextu, ale upozorňuje, že pro screening zcela zdravých jedinců prozatím neexistuje dostatek důkazů. Jen 5 z 130 zahrnutých studií se zaměřovalo na tuto populaci. Klinická a ekonomická utilita WGS pro preventivní screening zdravých jedinců je prozatím předmětem probíhajícího výzkumu. Tento nedostatek důkazů je ale z velké části způsoben tím, že donedávna bylo WGS příliš nákladné pro plošné nasazení. Situace se ale dynamický mění, přičemž náklady na sekvenování jednoho genomu se aktuálně pohybují na úrovni 500 EUR a náklady na přípravu sekvenačních knihoven pro WGS jsou dnes ve srovnání s cílenými panely nebo WES nižší.
Shrnutí
Genetické testy se liší především tím, na co se zaměřují a co jsou schopné vidět. Panelové sekvenování je efektivní pro jasně definované klinické otázky, WES rozšiřuje záběr na kódující oblasti celého genomu, ale WGS je jedinou metodou, která nedělá kompromisy a vidí na celý genom. Pro třídy variant, jakými jsou strukturní varianty, expanze STR a varianty v regulačních nebo hlubokých intronových oblastech, WGS konzistentně překonává WES. Klinická data ukazují, že právě tyto „neviditelné" varianty tvoří nezanedbatelnou část nevyřešených genetických diagnóz. S klesajícími náklady na sekvenování a rostoucí snahou o standardizaci klinických postupů, ekonomické i praktické argumenty pro WGS jako testu první volby postupně sílí.
Zdroje
- Austin-Tse CA et al. (2022). Best practices for the interpretation and reporting of clinical whole genome sequencing. npj Genomic Medicine, 7:27. DOI: 10.1038/s41525-022-00295-z
- Souche E et al. (2022). Recommendations for whole genome sequencing in diagnostics for rare diseases. European Journal of Human Genetics, 30:1017–1021. DOI: 10.1038/s41431-022-01113-x
- Pagnamenta AT et al. (2023). Structural and non-coding variants increase the diagnostic yield of clinical whole genome sequencing for rare diseases. Genome Medicine, 15:94. DOI: 10.1186/s13073-023-01240-0
- Dimmock D et al. (2018). Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. npj Genomic Medicine, 3:27. DOI: 10.1038/s41525-018-0053-8
- Chintalaphani SR et al. (2021). An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathologica Communications, 9:98. DOI: 10.1186/s40478-021-01201-x
- Boivin M & Ravenscroft G (2025). Current understanding of skeletal muscle repeat expansion disorders. Current Opinion in Neurology, doi:10.1097/WCO.0000000000001394
- Kelley CP et al. (2021). Molecular mechanisms underlying nucleotide repeat expansion disorders. Nature Reviews Molecular Cell Biology, 22:589–607. DOI: 10.1038/s41580-021-00382-6
- McElwee F et al. (2026). The cost and cost-effectiveness of whole-exome and whole-genome sequencing: a systematic literature review. European Journal of Human Genetics. DOI: 10.1038/s41431-026-02146-2
- Kaschta D et al. (2025). Evaluating genome sequencing strategies: trio, singleton, and standard testing in rare disease diagnosis. Genome Medicine, 17:67. DOI: 10.1186/s13073-025-01516-7
- Zeuli R et al. (2024). Whole genome sequencing identifies elusive variants in genetically unsolved Italian inherited retinal disease patients. Human Genetics and Genomics Advances, 5:100314. DOI: 10.1016/j.xhgg.2024.100314
- O'Donnell-Luria A et al. (2022). Recommendations for clinical interpretation of variants found in non-coding regions of the genome. Genome Medicine, 14:9. DOI: 10.1186/s13073-022-01073-3
- Kaplun A et al. (2025). ONT in Clinical Diagnostics of Repeat Expansion Disorders: Detection and Reporting Challenges. International Journal of Molecular Sciences, 26:2725. DOI: 10.3390/ijms26062725





